
—— Samarbejdsstudie af Zhejiang CDC, Macro & Micro-Test og China CDC. Udgivet i Frontiers in Cellular and Infection Microbiology.
Studieoversigt
I maj 2026 udgav Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4.6) en artikel ledet af Zhejiang Provincial Center for Disease Control and Prevention (Zhejiang CDC) med bioinformatikteamet fra Beijing Macro & Micro-Test Bio-Tech Co., Ltd. og National Institute for Communicable Disease Control and Prevention (China CDC) som medforfattere. Undersøgelsen har titlen:
"Identifikation og fylogenetisk analyse af syv Brucella abortus-stammer i Zhejiang, Kina."

Denne undersøgelse repræsenterer den første systematiske, helgenombaserede fylogenetiske sporbarhedsanalyse af Brucella abortus (B. abortus) i Zhejiang-provinsen, Kina. Holdet analyserede syv isolater indsamlet fra 2015 til 2025 (fire stammer af menneskelig oprindelse og tre stammer af kvægoprindelse fra Jinhua, Quzhou og Ningbo). Resultaterne giver genomisk bevis for oprindelsen og transmissionsruterne for denne "nordlige dominerende art" i en atypisk sydlig epidemisk region i det østlige Kina.
Baggrund og betydning
Brucellose er en zoonotisk sygdom forårsaget af bakterier af slægten Brucella. Brucella abortus inficerer primært kvæg, men kan også forårsage sygdom hos mennesker. I Kina viser brucellose markant geografisk variation: den højeste forekomst forekommer i de nordlige provinser (f.eks. Indre Mongoliet, Shanxi, Heilongjiang). I modsætning hertil har de sydlige provinser, herunder Zhejiang, historisk set været domineret af Brucella melitensis, med meget få rapporterede tilfælde af B. abortus. Denne regionale forskel gør den genetiske karakterisering og kildesporing af B. abortus i Zhejiang til en central folkesundhedsprioritet.
Metoder og nøgleresultater
Forskerholdet valgte en flerstrenget strategi, der kombinerede molekylærbiologi og bioinformatik:
1.Identifikation af patogener og grundlæggende typning
BCSP-31-gen-PCR og AMOS-PCR bekræftede, at alle syv isolater var B. abortus.
Multilocus-sekvenstypning (MLST) baseret på ni housekeeping-gener viste, at alle isolater tilhørte sekvenstypen ST2, hvilket indikerer høj genetisk homogenitet blandt de cirkulerende B. abortus-stammer i Zhejiang.
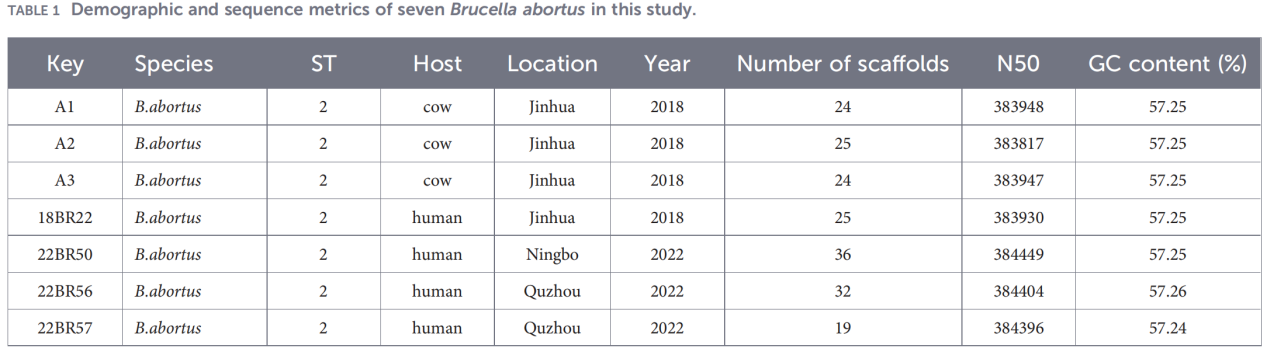
2.Helgenomkarakterisering
Helgenomsekventering blev udført på Illumina NovaSeq-platformen. Analyse af gennemsnitlig nukleotididentitet (ANI) viste, at Zhejiang-isolaterne havde op til 99,99 % lighed med referencestammen B. abortus 544.
Pan-genomanalyse afslørede en stærkt bevaret population: 3.084 kernegener blev identificeret, sammen med kun 10 skalgener, og ingen bløde kerne- eller skygener blev detekteret.
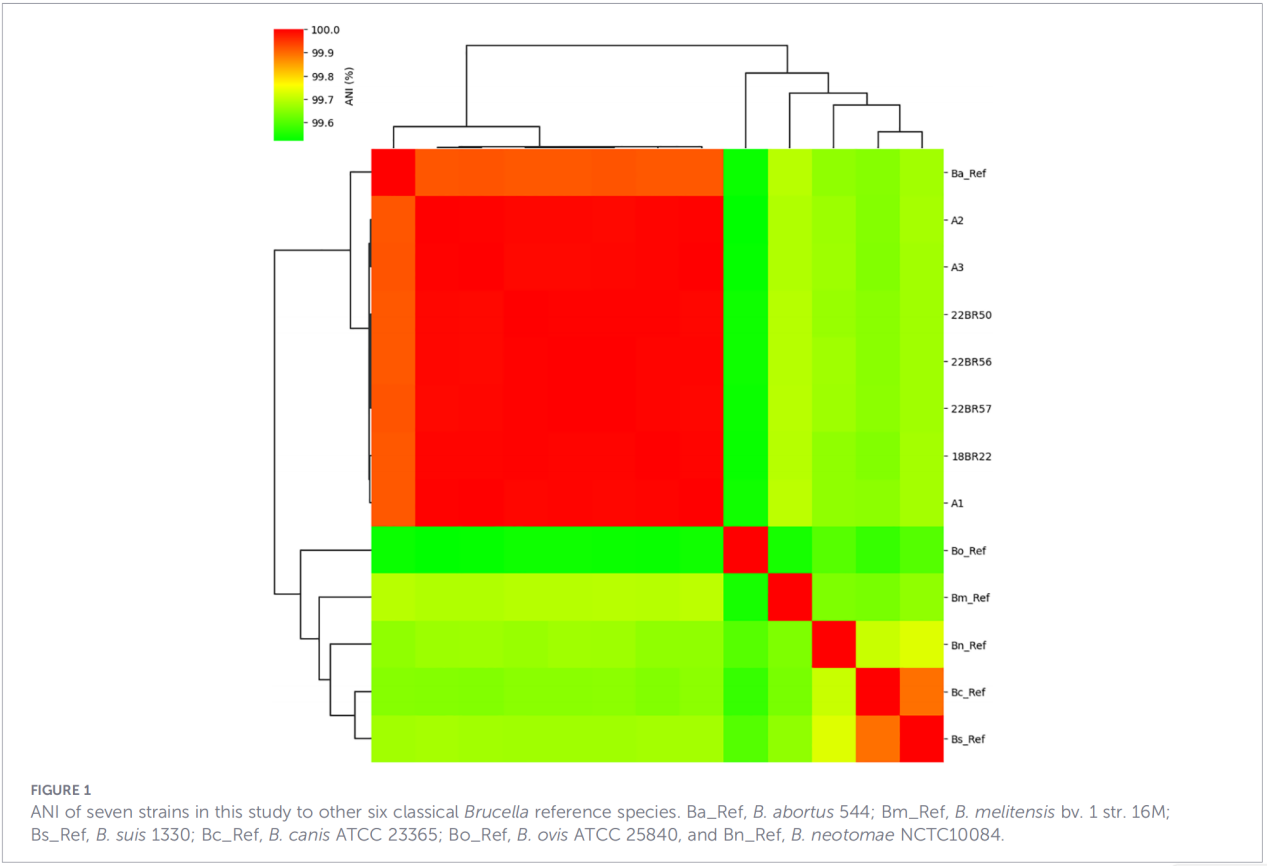
3.Genprofiler for virulens og antimikrobiel resistens
I alt 68 virulensrelaterede faktorer blev forudsagt, dækkende klassiske veje såsom LPS-biosyntese, T4SS-sekretionssystemet og BvrR-BvrS to-komponent reguleringssystem. Bemærkelsesværdigt manglede alle isolater adhæsingenerne bmaA og btaF. Resistensgenanalyse påviste kun mprF-genet i CARD-databasen, uden andre resistensdeterminanter identificeret.
 4. Fylogenetisk rekonstruktion og transmissionssporing
4. Fylogenetisk rekonstruktion og transmissionssporing
Core-genome single-nukleotid polymorphism (cgSNP) analyse placerede Zhejiang-isolaterne på en specifik position i det globale fylogenetiske træ. Resultaterne viste, at Zhejiang-stammerne danner en monofyletisk gruppe sammen med stammer fra Rusland, Mongoliet og flere nordlige kinesiske provinser (Ningxia, Heilongjiang, Indre Mongoliet, Hebei, Gansu, Beijing). Denne gruppe opdeles yderligere i tre forskellige underklader (klade 1-3), hvilket tyder på flere uafhængige introduktionsbegivenheder.
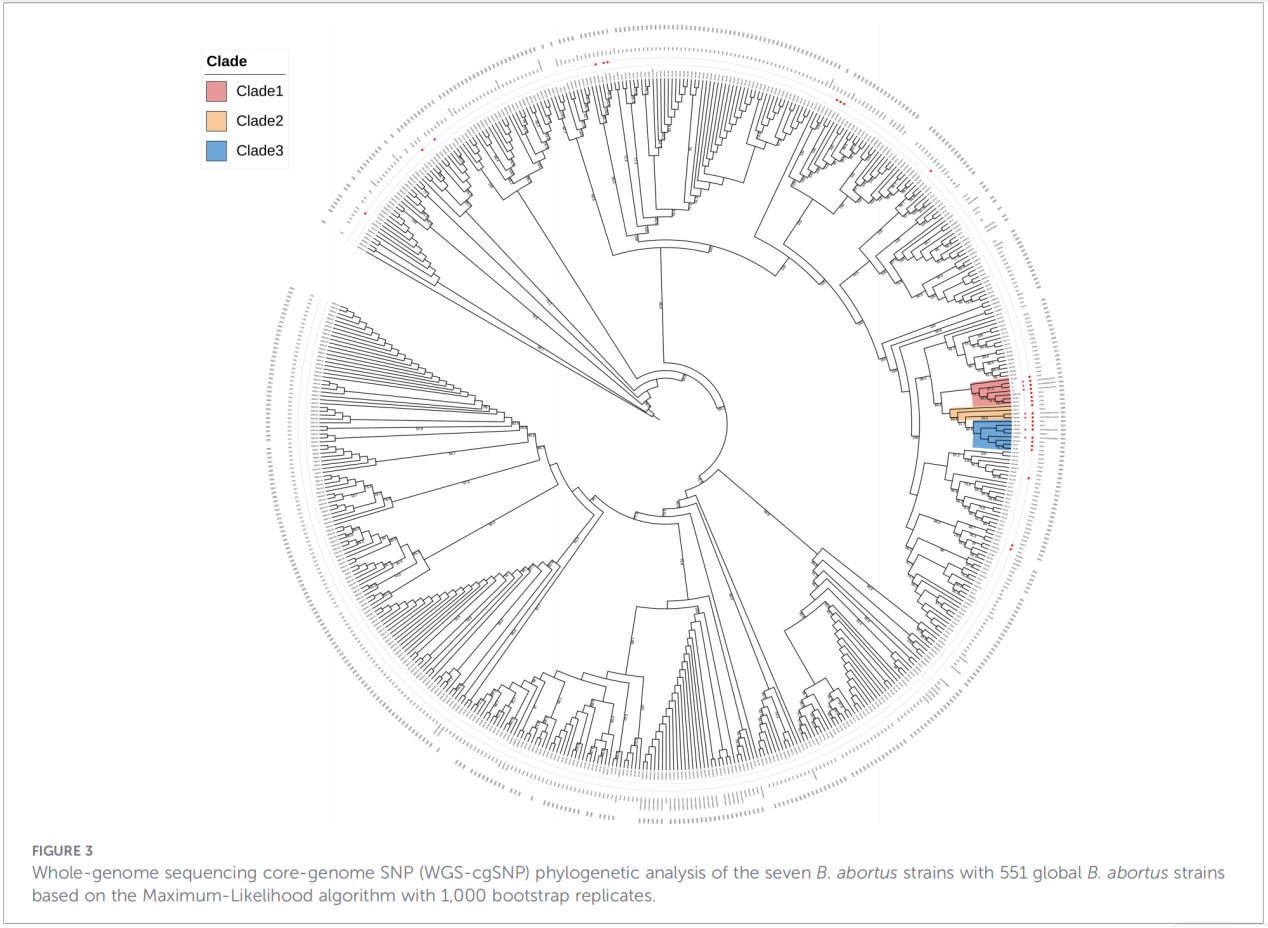
Konklusioner og implikationer
Denne undersøgelse leverer det første højpræcisions genomiske datasæt for B. abortus i Zhejiang-provinsen og giver flere vigtige konklusioner:
- Clear genetisk baggrund– B. abortus-stammerne, der cirkulerer i Zhejiang, tilhører ST2, er genomisk stærkt konserverede og repræsenterer en typisk bovin brucellose-afstamning.
2. Evidensitet af tværregional transmission– Fylogenetisk analyse understøtter ikke eksistensen af en uafhængig endemisk afstamning i Zhejiang. I stedet tyder dataene stærkt på, at disse stammer stammer fra det nordlige Kina og muligvis deler en fælles evolutionær baggrund med stammer fra Rusland og Mongoliet. Tilstedeværelsen af tre underklader antyder flere separate introduktionsbegivenheder.
3. Folkesundhedsmæssige konsekvenser– Resultaterne understreger værdien af genomisk overvågning af brucellose, selv i traditionelt ikke-endemiske regioner som f.eks. Zhejiang. Selvom det nuværende antal tilfælde er lavt, kan højopløsningsværktøjer som cgSNP effektivt spore kilden til importerede udbrud og give videnskabelig dokumentation for at afbryde smittekæder forbundet med transport af husdyr mellem provinsielle stater.
Dette arbejde udfylder ikke blot et forskningshul i Zhejiang-provinsen, men leverer også nye basisdata til patogenovervågning og risikovurdering af brucellose i Yangtze-floddeltaregionen.
Papirinformation:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Identifikation og fylogenetisk analyse af syv Brucella abortus-stammer i Zhejiang, Kina. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Opslagstidspunkt: 10. juni 2026